Fibrodysplasia Ossificans Progressiva (FOP), often called the “stone man disease,” is one of the rarest and most astonishing genetic disorders known to medicine.
It causes the body’s soft connective tissues—muscles, tendons, and ligaments—to slowly transform into bone through a process called heterotopic ossification.
Over time, this extra bone fuses joints and creates a rigid “second skeleton,” locking the body in place until movement becomes nearly impossible.
The root cause lies in a mutation of the ACVR1 gene, which controls bone growth signaling.
This tiny genetic error keeps the receptor perpetually active, turning everyday inflammation or minor injuries into instructions for new bone formation where it does not belong.
The bone that grows is real, mature bone—identical to the skeleton—but it appears in all the wrong places, gradually replacing flexible tissue.
Symptoms frequently begin in early childhood with a distinctive clue: malformed big toes.
Many babies are born with short, inwardly turned great toes or missing joints, a congenital marker that often goes unnoticed until flare-ups start.
These painful swellings can erupt spontaneously or after something as simple as a bump or injection.
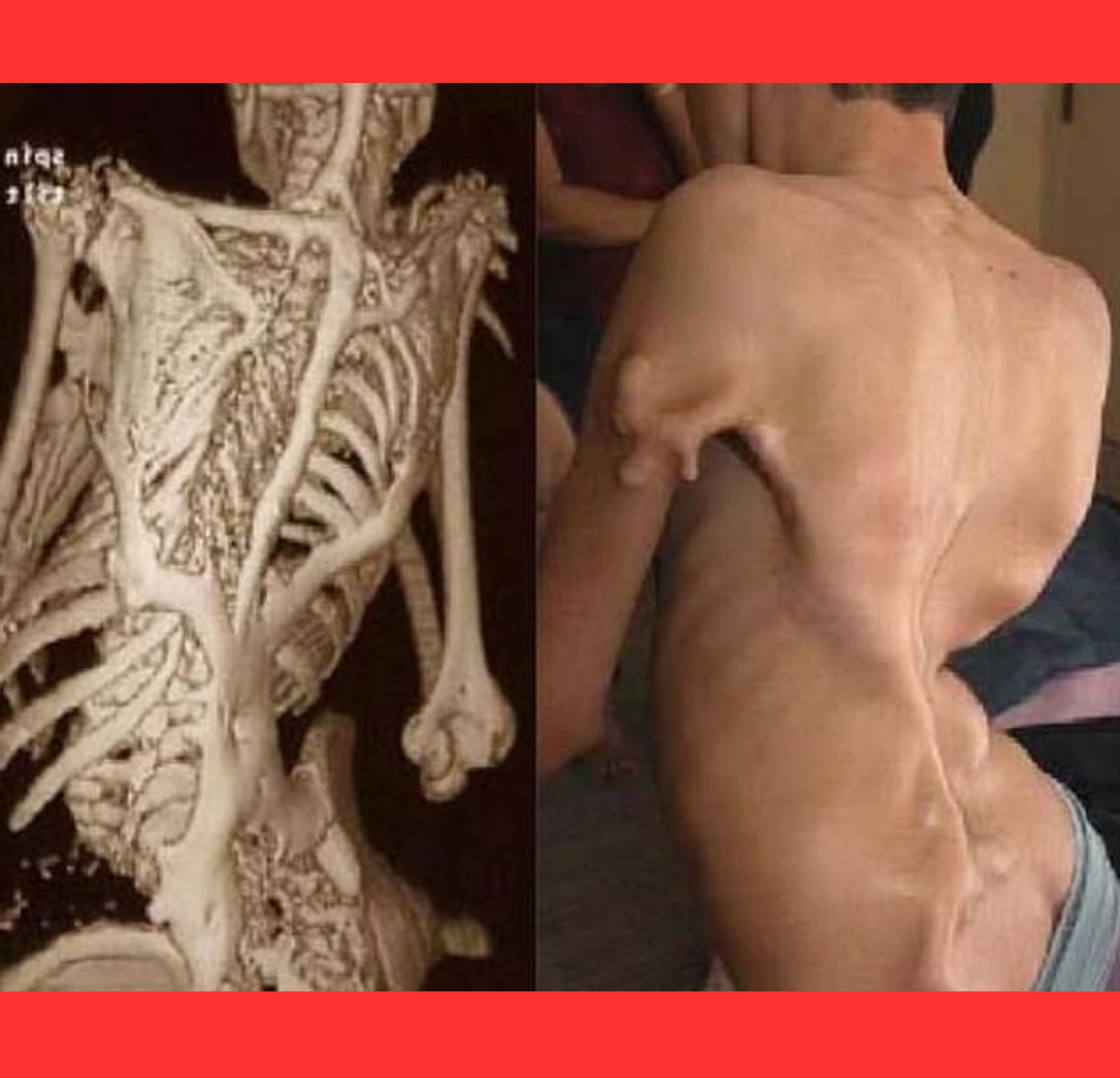
During a flare-up, soft tissue inflames, swells into a tumor-like mass, and eventually hardens into bone over weeks or months.
Once formed, the new bone cannot be removed without triggering even more growth.
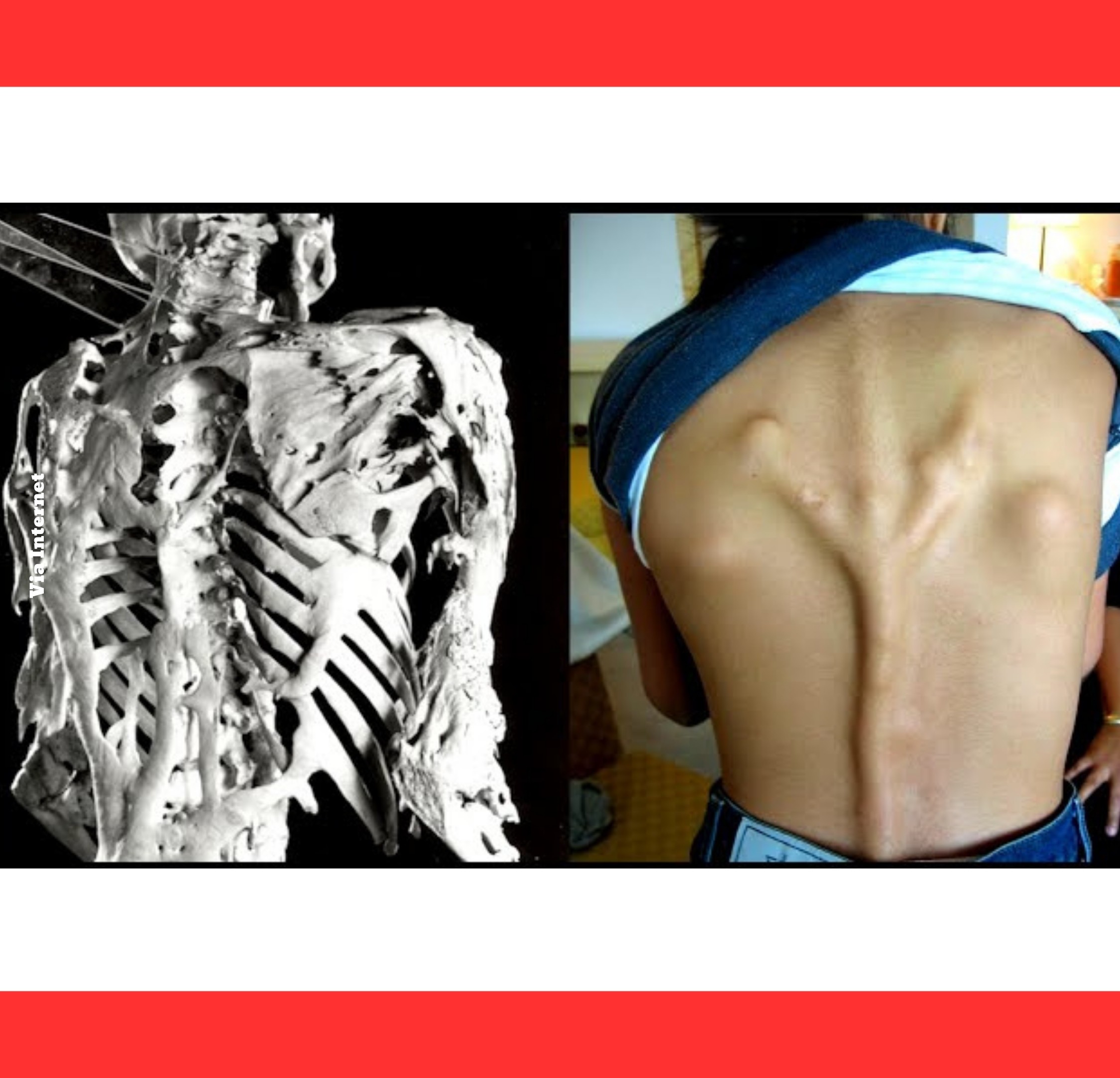
The ossification spreads in a predictable pattern—starting at the neck and shoulders, moving downward through the torso and limbs—mirroring the way a fetus develops its skeleton.
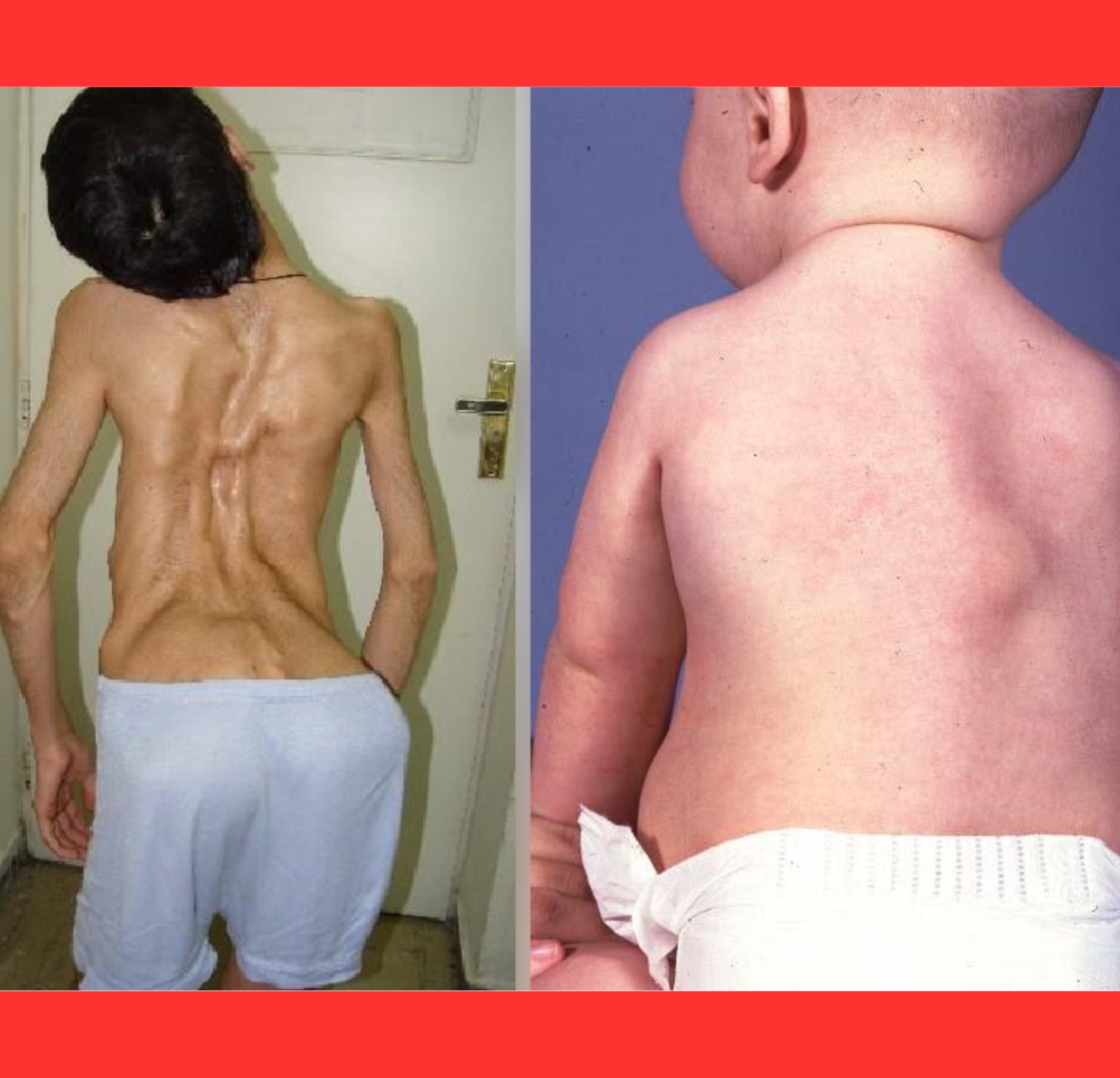
As the second skeleton expands, joints lock permanently. Shoulders freeze, the spine stiffens, and hips or knees become immobile.
Simple actions like walking, dressing, or turning the head grow increasingly difficult.
By early adulthood, most patients rely on wheelchairs; many become almost completely rigid, yet their minds remain sharp and alert.
The disorder spares only a few areas, such as the diaphragm, tongue, and heart muscle, but even these offer limited relief.
Rib-cage restriction often leads to breathing difficulties, while jaw fusion can make eating and speaking a challenge.
Pneumonia and respiratory failure are common complications, shortening life expectancy to around 40 years without careful management.
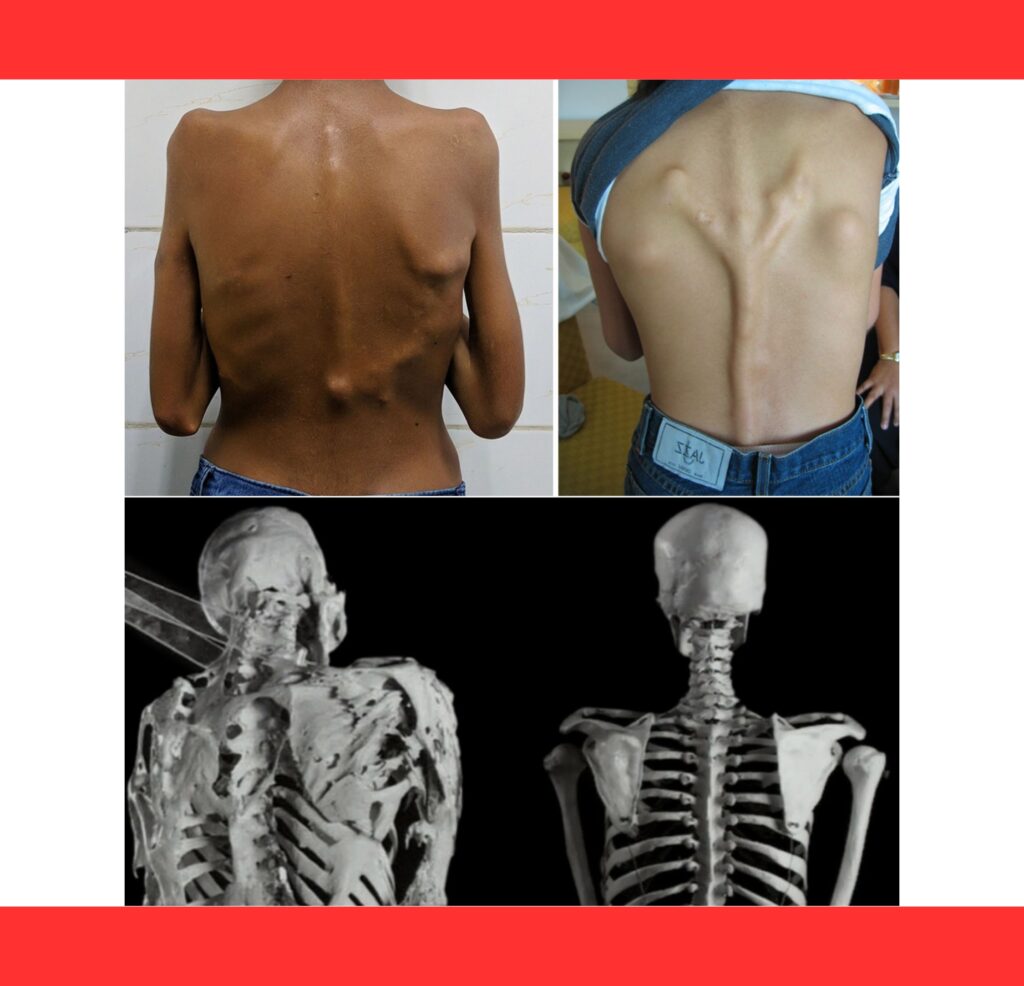
FOP affects roughly one in two million people worldwide, striking all ethnicities equally.
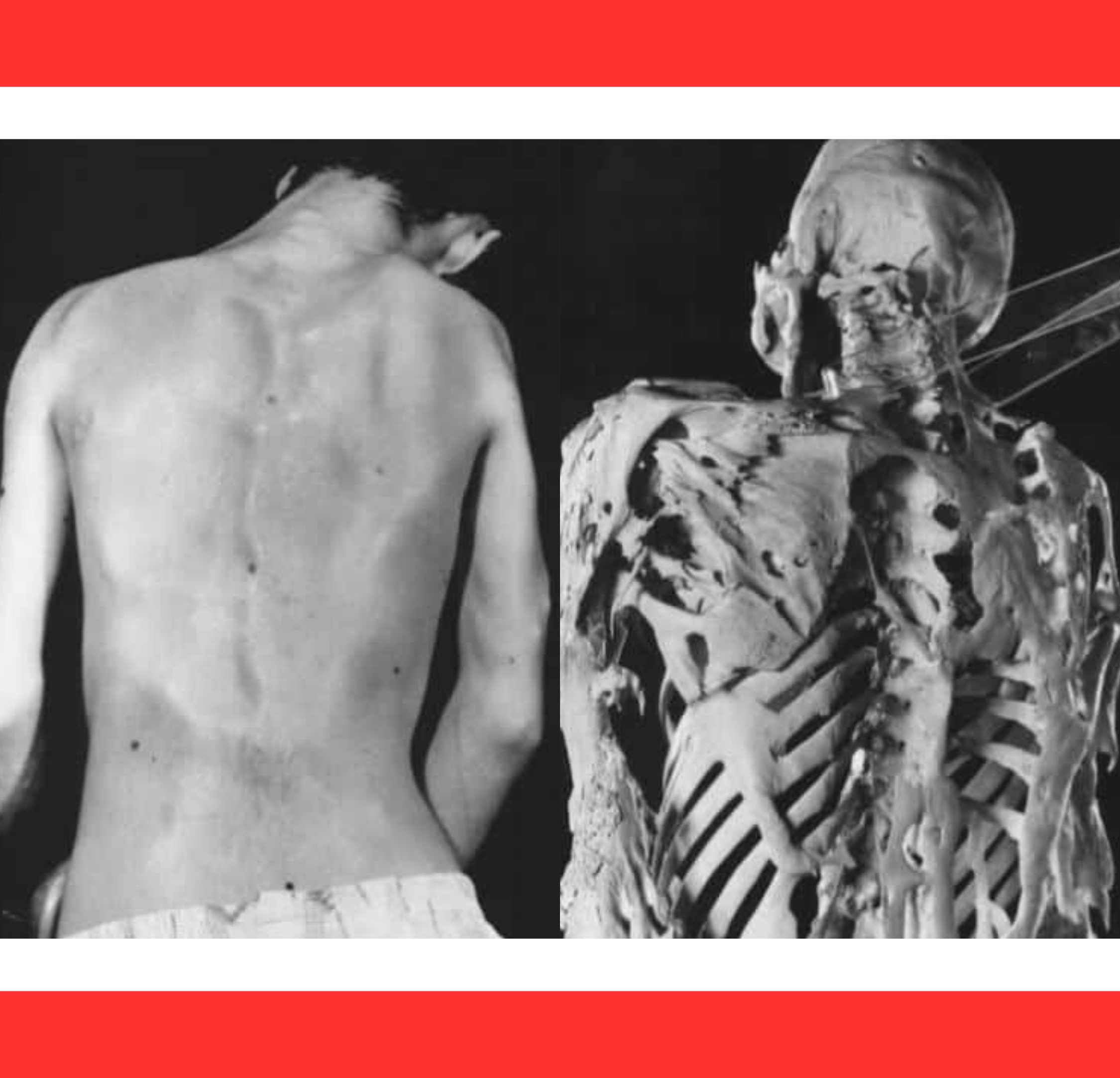
First described in 1648 and later detailed through cases like Harry Eastlack, whose fully ossified skeleton is preserved in Philadelphia’s Mütter Museum, the disease has haunted medical history for centuries. Fewer than 1,000 confirmed cases exist today.
Diagnosis combines clinical signs, imaging, and genetic testing. X-rays reveal sheets of extra bone bridging joints, while early blood work and MRI can catch pre-bone swelling.
Misdiagnosis as cancer or injury often leads to harmful biopsies that accelerate ossification, underscoring the need for awareness among doctors.
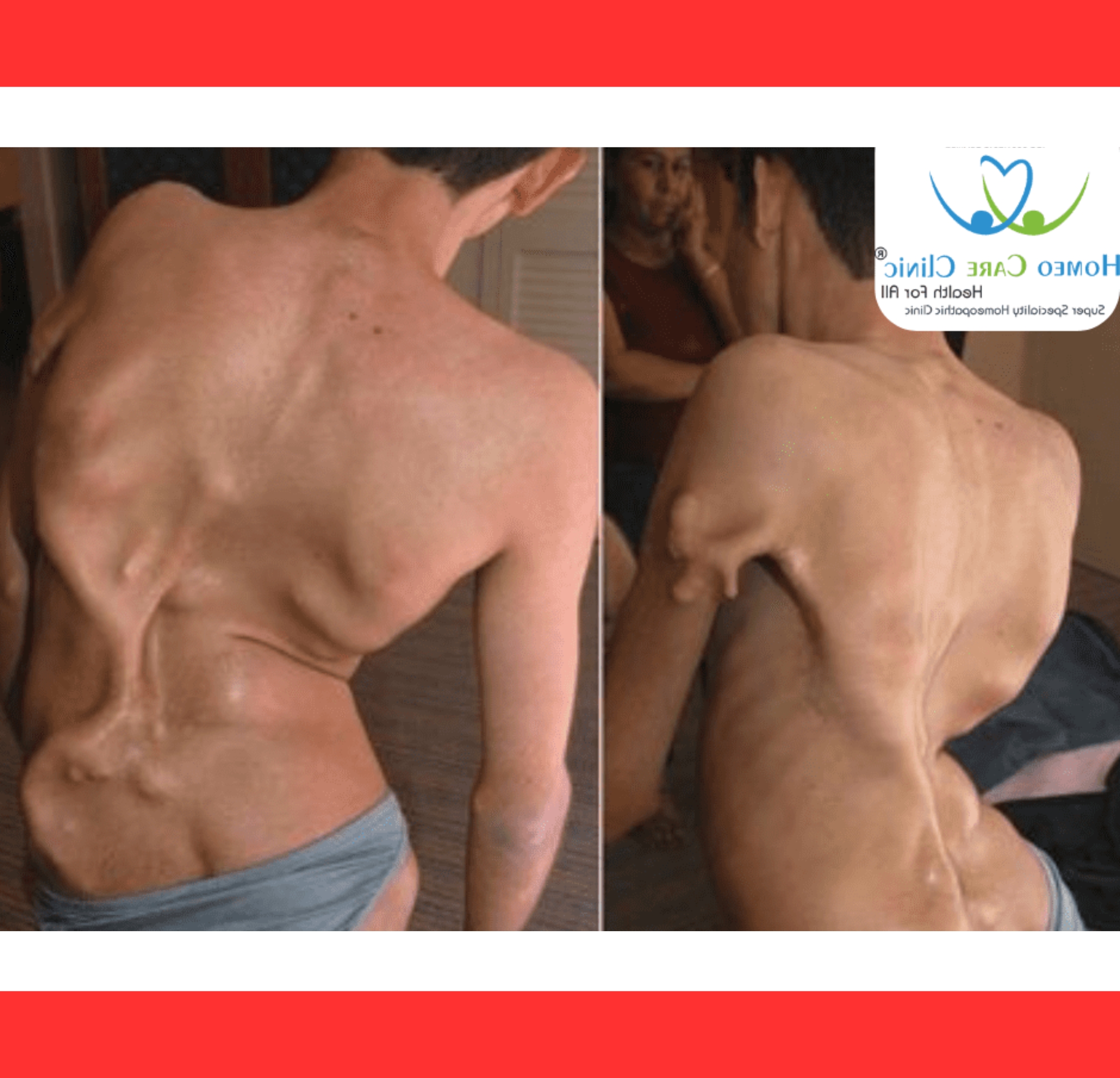
There is no cure, so treatment focuses on prevention.
Patients must avoid trauma, falls, intramuscular injections, and unnecessary surgery, as any injury can spark explosive bone growth.
Anti-inflammatory drugs help tame flare-ups, and physical therapy is kept gentle to maintain whatever mobility remains.
Recent breakthroughs offer cautious hope.
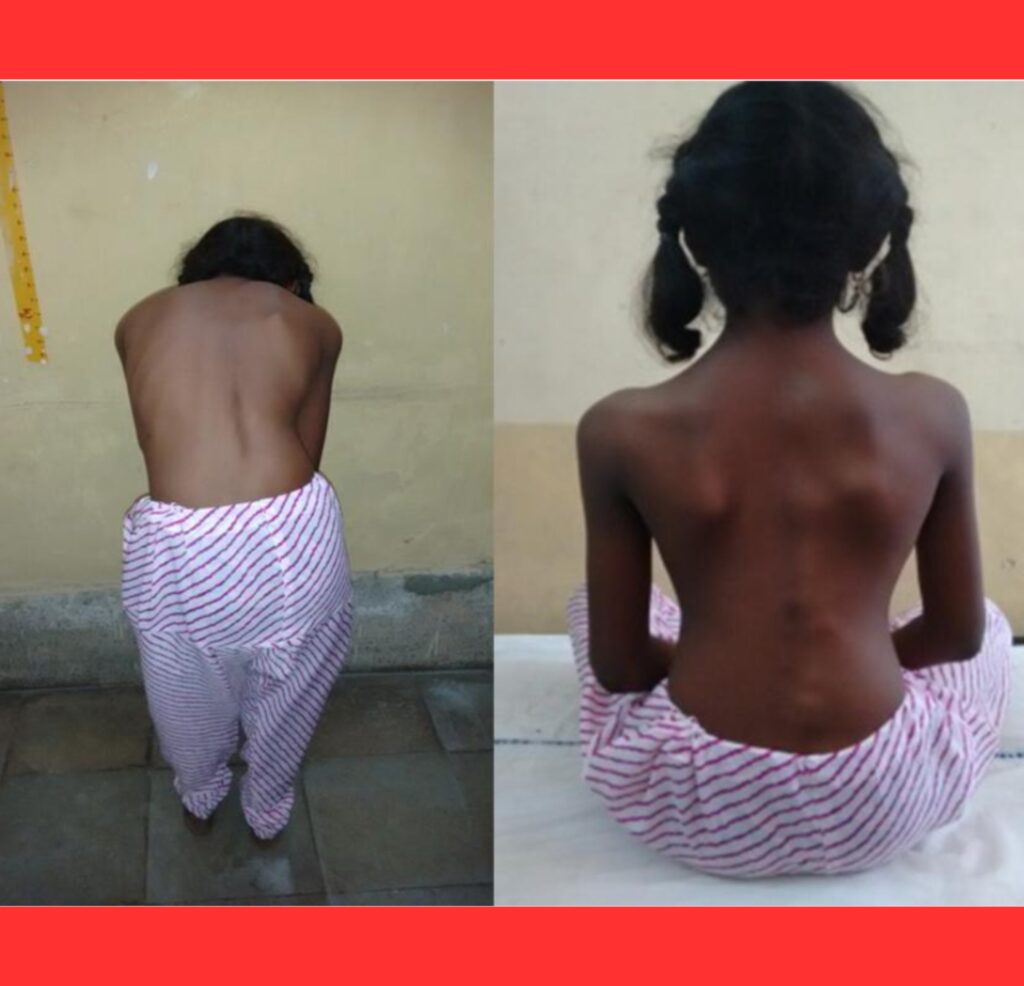
The FDA-approved drug palovarotene (Sohonos) reduces new bone formation by about 50-60 percent in clinical trials.
Other experimental therapies targeting the ACVR1 pathway, including kinase inhibitors and antibodies like garetosmab, are advancing through trials, raising the possibility of slowing or even halting progression.
Though FOP remains a cruel and relentless condition, ongoing research and global patient networks bring renewed optimism.
Families and scientists continue to push boundaries, determined that one day the body’s tragic urge to turn flesh to stone may finally be silenced.
Until then, awareness and careful care remain the best shields against this extraordinary disorder.