In the vast landscape of rare medical conditions, Olmsted syndrome emerges as a particularly debilitating genodermatosis.
This ultra-rare disorder disrupts normal skin keratinization, resulting in thick, callus-like growths primarily on the palms, soles, and periorificial areas including the mouth.
First described in 1927, it affects an estimated fewer than 100 individuals worldwide, making diagnosis and treatment exceptionally challenging for patients and physicians alike.
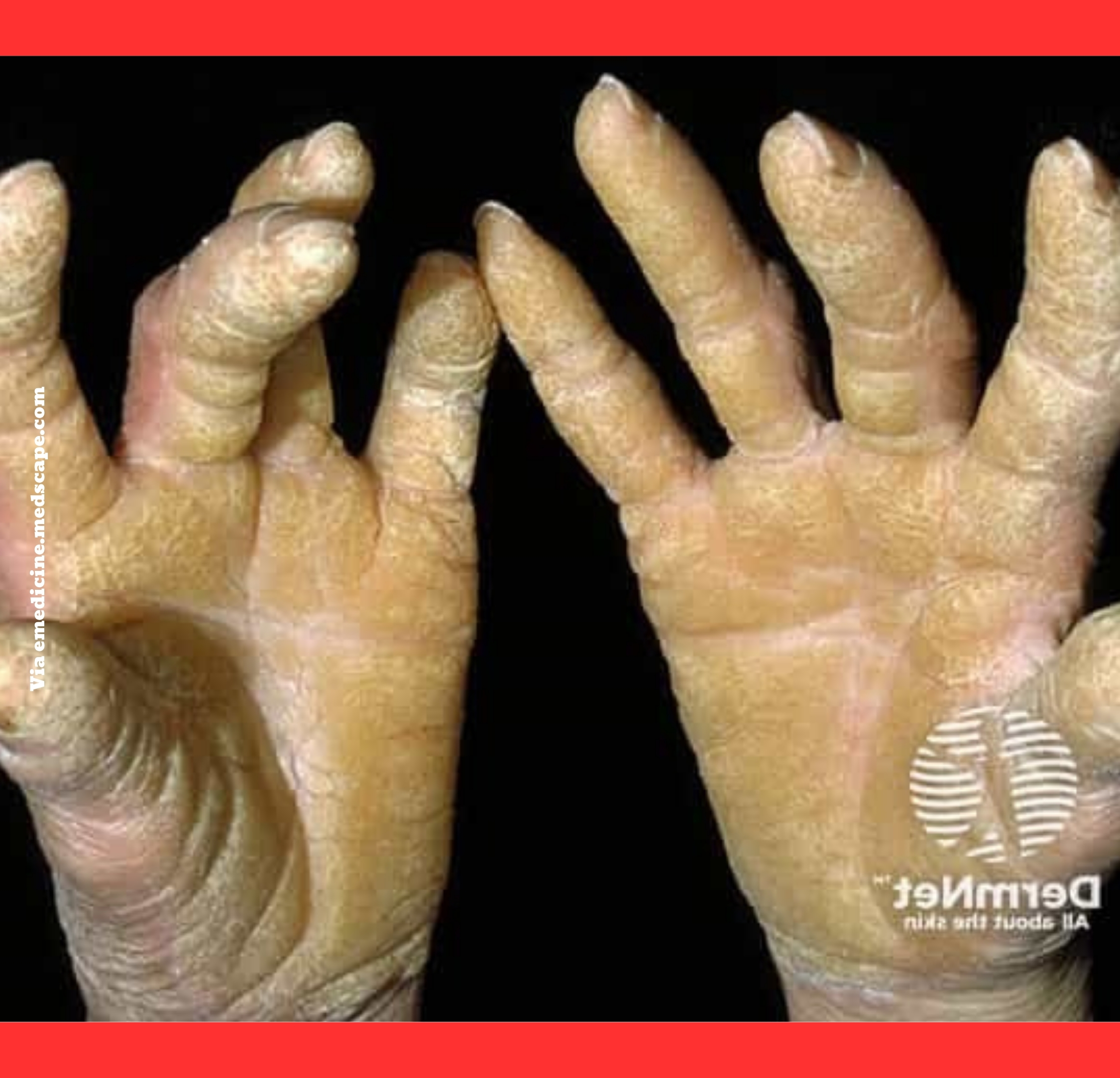
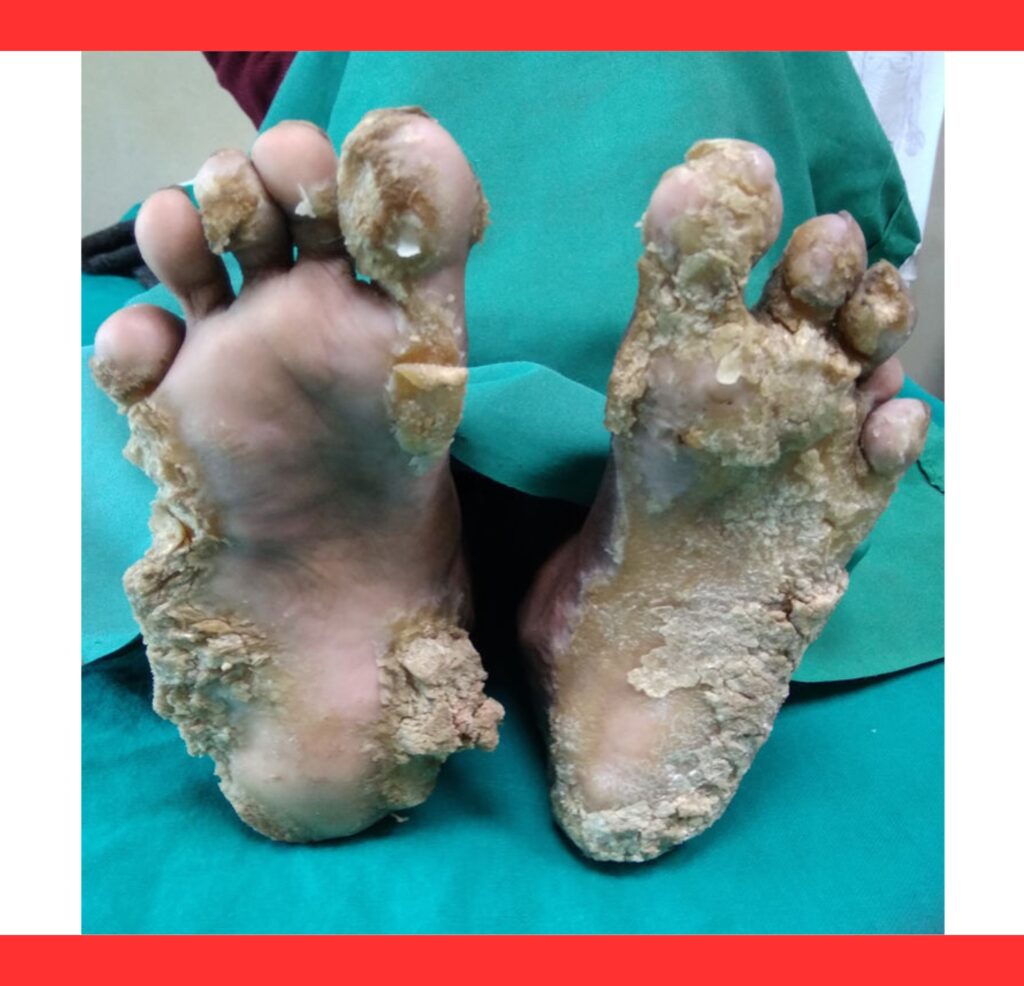
The most striking feature is bilateral palmoplantar keratoderma.
Affected individuals develop rigid, yellowish, fissured hyperkeratotic plaques on their hands and feet.
These calluses cause intense pain, cracking, and bleeding, severely impairing daily functions like walking or using tools.
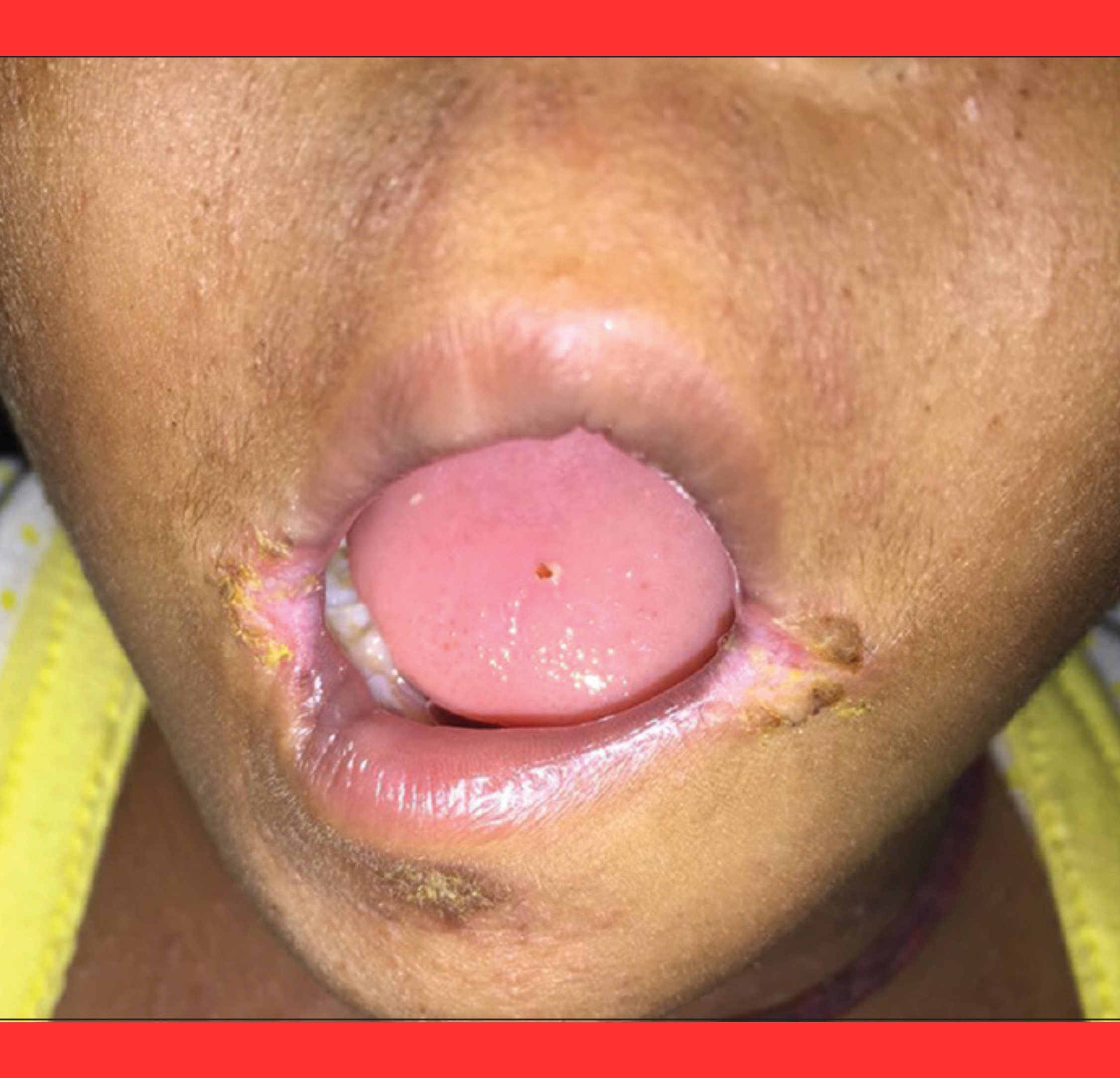
Compounding the physical toll, periorificial keratotic plaques often form around the mouth and other orifices.
These lesions create crusty, thickened areas that interfere with eating, speaking, and breathing, leading to significant discomfort and social challenges.
Symptoms of Olmsted syndrome usually begin in infancy or early childhood and progress relentlessly.
The hyperkeratosis extends beyond typical boundaries, creating a transgredient pattern that exacerbates functional limitations over time.
Families often face years of uncertainty before receiving an accurate diagnosis.
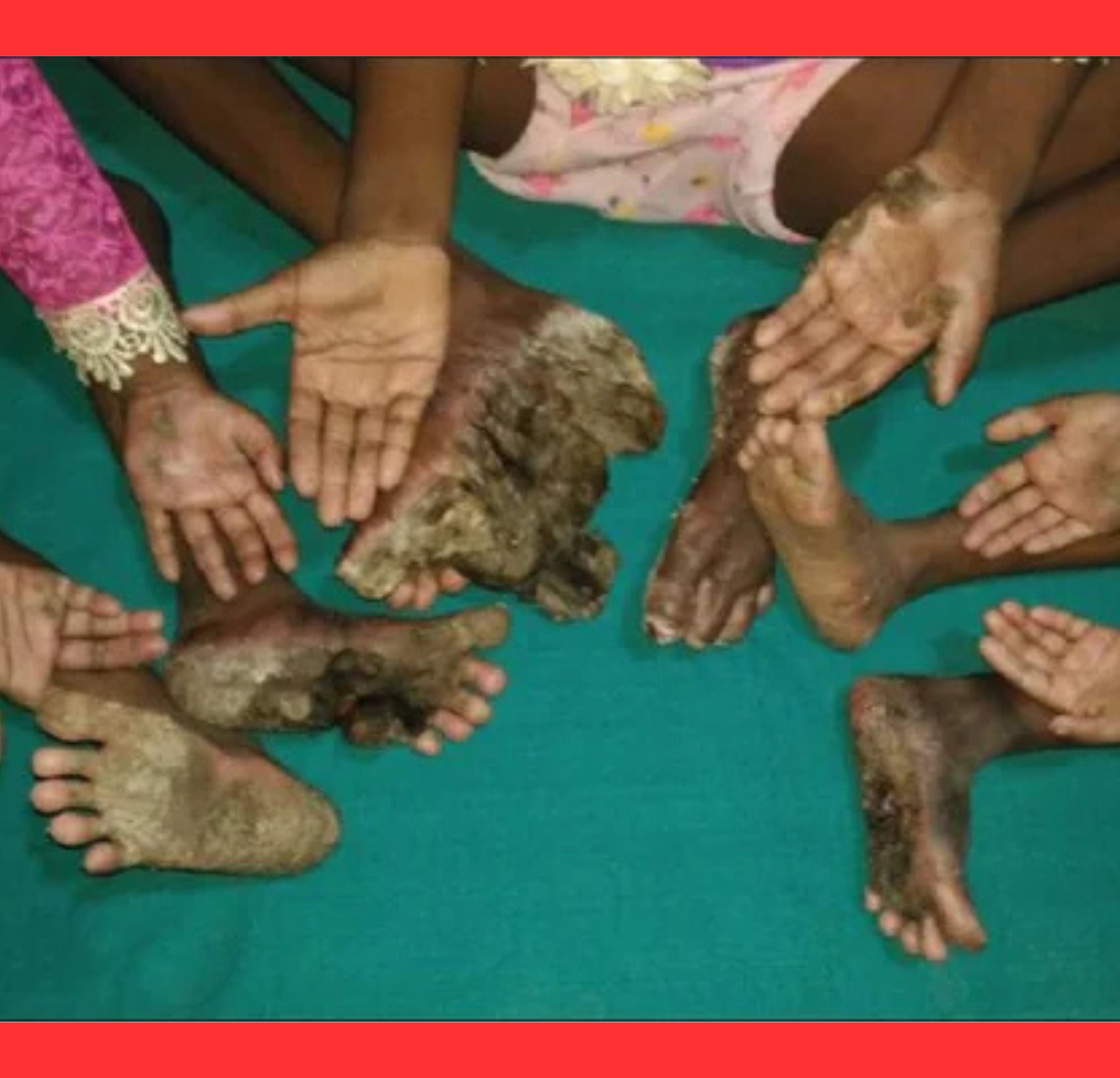
At its core, the condition stems from gain-of-function mutations in the TRPV3 gene on chromosome 17.
This gene regulates calcium channels in skin cells, and mutations trigger excessive keratinocyte proliferation and abnormal skin thickening.
Some rarer cases involve mutations in the MBTPS2 gene.
Diagnosing Olmsted syndrome proves challenging due to its rarity.
Clinicians rely on characteristic clinical signs, skin biopsies showing marked hyperkeratosis, and confirmatory genetic testing to differentiate it from similar disorders like Vohwinkel syndrome or other palmoplantar keratodermas.
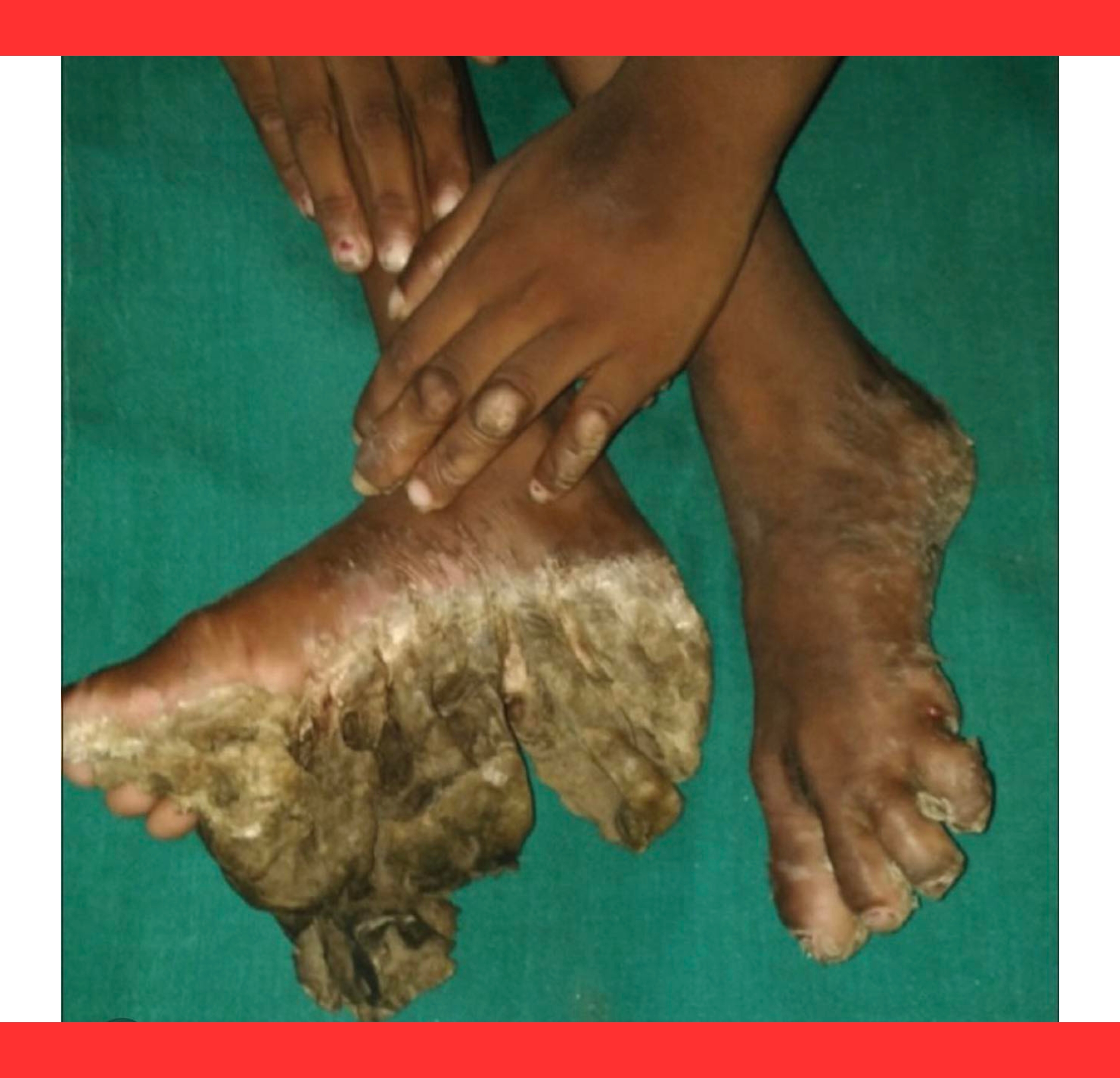
Complications can be mutilating and life-altering.
Constricting bands known as pseudoainhum may encircle digits, causing flexion deformities, infections, or even autoamputation.
Chronic pruritus, pain, and secondary infections further diminish quality of life for those living with the condition.
Currently, no cure exists for Olmsted syndrome.
Standard management involves symptomatic relief with emollients, keratolytics such as urea or salicylic acid preparations, and oral retinoids to soften and reduce the hyperkeratotic plaques.
Surgical interventions may be needed in severe cases to restore function.
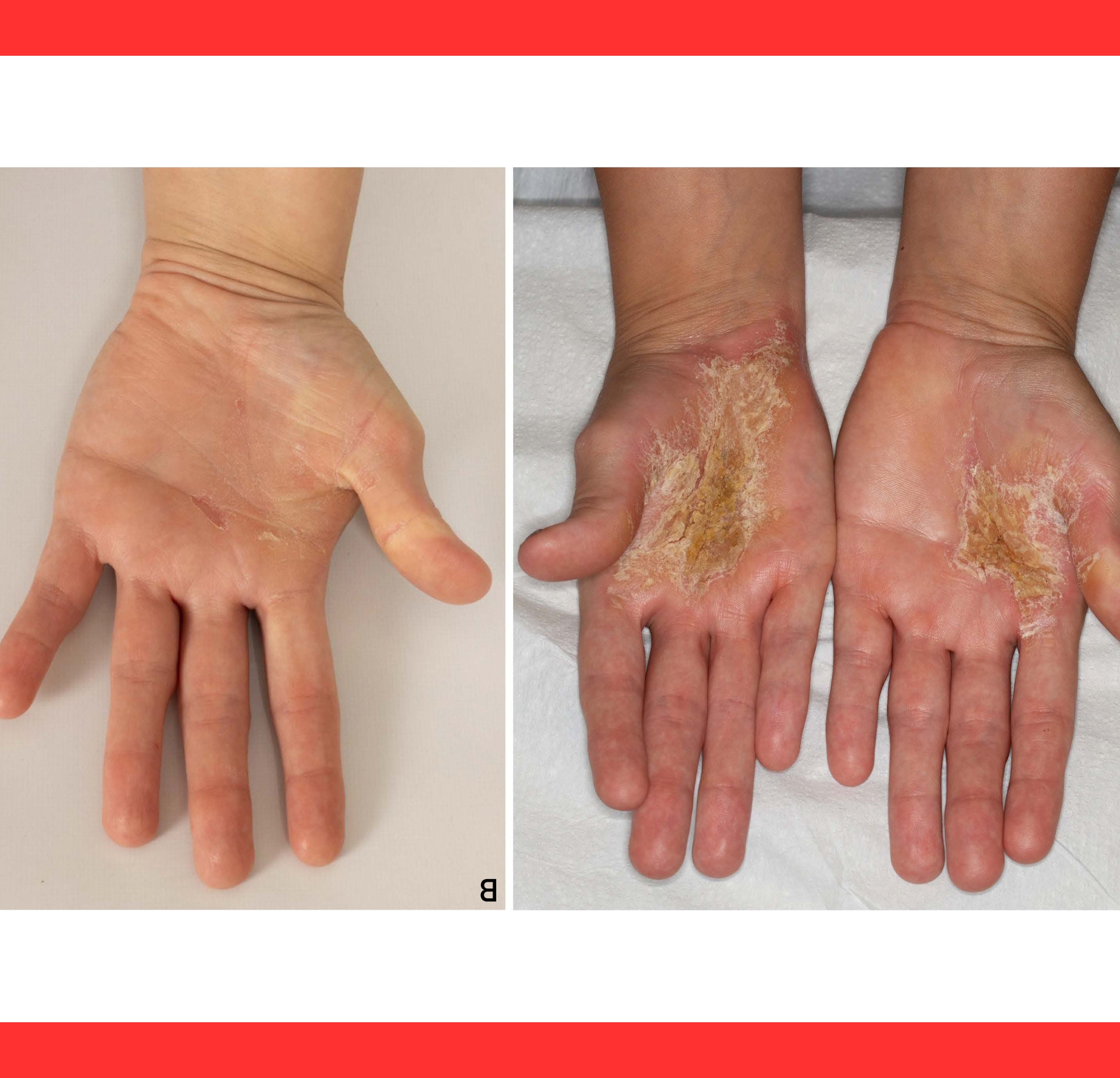
Exciting progress in targeted therapies offers new hope.
EGFR inhibitors like erlotinib have demonstrated remarkable efficacy in select patients, leading to substantial plaque reduction and improved mobility and comfort.
Ongoing research into gene therapies continues to advance.
For those living with Olmsted syndrome, the daily struggle demands resilience and multidisciplinary support.
Patient stories reveal profound impacts on physical, emotional, and social well-being, underscoring the importance of comprehensive care and community resources from dermatologists, geneticists, and psychologists.
Raising awareness about Olmsted syndrome is crucial for early diagnosis and research funding.
As scientists pursue advanced genetic and molecular treatments, there is optimism for transformative breakthroughs that could one day alleviate the suffering of affected individuals and their families.